厉害了!董广彬一天两篇Nature大子刊!
第一篇


▲第一作者:: Jun Zhu, Rui Zhang
通讯作者:Guangbin Dong
通讯单位:University of Chicago
DOI: 10.1038/s41557-021-00757-4
01
背景介绍
烯烃复分解反应(olefin metathesis)虽然已经被应广泛应用于有机合成和材料科学,但碳-碳单键的直接复分解却极为罕见。
02
本文亮点
u 本文报道了一种钌催化的可逆C-C单键复分解反应,允许氧化还原和pH中性的联芳基合成。
u 在导向基团的辅助下,非应变的同型联芳基化合物在空气稳定的钌(II)配合物的催化下,进行芳基交换以生成交叉联芳基产物。
u 在典型的交叉偶联反应下,反应的官能团(如卤素、硅基和硼酸盐部分)在复分解条件下是相容的。
u 机理研究表明,该反应途径涉及一个7配位、18电子闭壳中间体。

03
图文解析
钌催化烯烃复分解的一般机理包括催化剂活化、底物配位、π键裂解和重组、底物分解和催化剂再生等步骤(图1a);作者假设以Ru(II)-Ar作为催化剂/引发剂,首先联芳基底物与Ru配位,进行氧化加成,裂解C(芳基)-C(芳基)键(图1b);三芳基Ru配合物随后的还原消除可导致新的C(芳基)-C(芳基)键形成,并重新形成Ru(II)-Ar物种,该物种可以通过类似的顺序与另一个联芳基底物反应。因此,可以实现两种联芳基之间的C-C单键复分解(图1c)。

▲图1 碳-碳双键和单键的复分解反应
模型研究和DFT计算。为了验证这一假设,作者使用了两个联芳基底物(1a和2a)作为模型底物(图2a)。Ru(II)配合物(cat 1)包含一个类似于底物单体的芳基配体。在对各种反应条件进行了广泛的调查之后,得到了期望的复分解产物(3a),尽管其收率只有17%。为了更好地理解C-H和C-C激活途径之间的竞争,作者进行了密度泛函理论(DFT)计算。这两种途径的能量分布如图2b所示。

▲图2 钌催化联芳基复分解反应的探索
反应优化。C4取代的底物1b和2b,在Cat4的催化下复分解反应增强(表1)。
表1反应优化

反应机理。在优化条件的基础上,作者进一步探讨了反应机理。反应24h后,图3的两个反应得到了比例非常相似的同型二聚体和交叉二聚体的混合物。这些结果支持了复分解反应是可逆的,可以达到反应平衡的结论。此外,在标准条件下对模型反应的动力学监测表明,该反应没有诱导期。

▲图3 机理研究
底物拓展。表2研究了底物可以耐受哪些替代官能团,以及哪些DGs可以更有效。结果表明,在苯基C4/4’位置上的空间位阻确实可以抑制不需要的C-H活化途径。
表2联芳基复分解反应的底物范围

反应范围和局限性。芳基溴化物和硼酸酯是各种交叉偶联反应中最常见的反应部分,因此能够同时耐受溴化物和硼酸酯官能团的交叉二芳基合成极为罕见。双芳基复分解法的正交反应性可从双溴和双吡那氯硼烷底物的交叉复分解中看出(图4a)。含有二芳基1aa的吡唑与化学计量比的cat 7和AgOPiv反应,以40% NMR产率提供芳基转移产物3am(图4b)。此外,在较高温度下,观察到联芳基1aa和吡啶基底物2b之间的交叉复分解反应。目前正在努力了解各种DG之间的反应性差异,并通过更好的催化剂设计提高与非吡啶DG的反应性。

▲图4 反应范围和局限性
第二篇

▲第一作者:Xukai Zhou
通讯作者:Yan Xu ,Guangbin Dong
通讯单位:University of Chicago,California Institute of Technology
DOI: 10.1038/s41929-021-00661-7
01
背景介绍
芳烃环和杂芳烃稠环普遍存在于药物、天然产物和其他生物活性化合物的结构中。芳香环上C-H键和其环上烷基链之间的直接环化是获取这种结构非常便捷的方法。但是,由于需要特殊的反应基团、反应条件苛刻,这种环化策略受到了极大的阻碍。
02
本文亮点
◆ 本文报道了一种从线性简单酮前体制备多种芳香稠环的脱酰基环化策略,具有广泛的官能团耐受性。
◆ 反应机理:该反应通过前芳香族中间体均裂酮αC-C键,然后进行自由基介导的脱氢环化。

03
图文解析
芳烃和杂芳烃熔合环在药物、天然产物和其他生物活性化合物的结构中很常见(图1a);C-H烷基环化,提供了一种直接获得这些稠环的方法,其可以避免芳烃的预功能化(图1b);如果可以实现烷基酮的位置选择性C–C裂解以生成用于环化的活性烷基末端,例如碳中心自由基,则可以从简单的芳烃和烷基底物得到两相环化策略(图1c)。
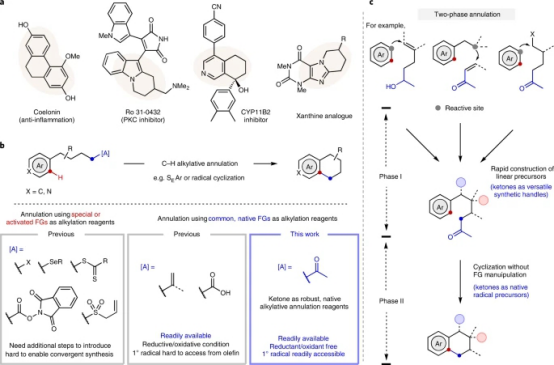
▲图1 芳香C(sp2)-H键的烷基环化反应
反应设计。在紫外光照射下,通过Norrish-Young反应实现了酮α C-C键的均裂断裂;也可以通过将酮转化为相应的肟酯/活化醚或叔醇,然后β-断裂来实现。作者前期报道了以一种铱催化裂解直链烷基酮的αC-C键的方法(图2)

▲图2 反应设计
反应发现与优化。为了验证该方法的可行性,作者选择5-(蒽基)戊烷-2-酮(1)作为模型底物来进行研究。从表1可发现(entry 1, entry 5),在该催化体系下,1能够以高产率转化为芳香稠环2。
底物拓展。在获取最佳反应条件后,接下来研究了酮的底物范围(表2)。从表2发现,该底物具有良好的兼容性且均能获得满意的产率。
机理研究。脱酰环化反应的详细机理尚不清楚,作者提出了较为可能的反应途径(图3a)。该反应可能从酮和肼D1形成腙开始,然后通过Ir催化的[3+2]环加成反应与1,3-二烯和烯烃迁移形成前芳香族中间体(Int 2);随后,可发生与Ir(I)催化剂的定向N-H氧化加成得到Ir(III)-氢化物中间体(Int 3),其随后经受芳构化驱动的均裂C-C裂解以形成吡唑配位Ir(II)氢化物(Int 4)和烷基(Int 5);Int 5经过自由基环化得到Int 6。

▲图3 反应机理
方法应用。作者进一步展示了脱酰基环化策略在芳香稠环的流线型两相合成中的应用(图4)。例如,双环二羧酸酯49(抗惊厥活性50的关键中间体),可通过本文的方法两步合成得到;然而,常规合成需要五步(图4a);黄嘌呤类似物54之前是通过三步合成,而本方法只需要两步且总收率加倍(图4b)。该两相环化策略还可进一步应用于N-杂芳烃基复杂多环化合物的后期构建(图4c)。

▲图4 反应的应用
作者介绍

董广彬
董广彬,本科毕业于北京大学,博士毕业于斯坦福大学,现为芝加哥大学教授。专长于有机合成,催化,有机金属和药物化学领域。
研究领域:
1) C–C Bond Activation. Our long-term goal is to greatly extend the regime of C−C activation to a wide range of organic compounds and enable synthetically useful transformations.
2) Byproduct-Free Ketone Alkylation.
3) Non-Directed β-Functionalization of Carbonyl Compounds.
4) Exo-type Directing Group for Site-selective C−H Activation.
5) Palladium/Norbornene Catalysis.
6) Total Synthesis and Drug Discovery.
7) Polymer Chemistry.
